This article delves into the groundbreaking research identified by 00957, exploring the potential of the gut microbiome as a predictive marker for clinical improvement in patients with Rheumatoid Arthritis (RA). We analyze a retrospective, observational cohort study that investigates the relationship between gut microbiota composition and clinical outcomes in RA patients, focusing on the Minimum Clinically Important Improvement (MCII).
Study Participants and Baseline Characteristics
The study meticulously examined data from 32 participants diagnosed with RA, carefully selected from a larger biobank of 86 patients. These individuals had at least two stool samples collected 6 to 12 months apart and complete clinical data from both visits. Patients in clinical remission at both visits were excluded to focus on those with active disease. The cohort was predominantly female (65.6%) with a mean age of 64.9 years and an average disease duration of 8.2 years, indicating established RA.
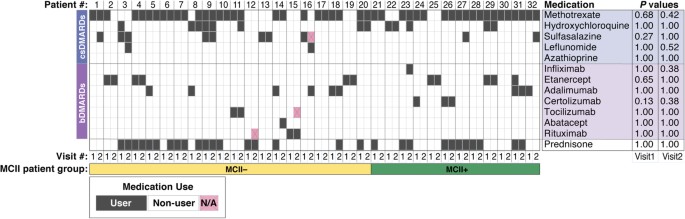
At the initial stool sample collection, all patients were undergoing treatment, including biologic disease-modifying anti-rheumatic drugs (bDMARDs, 46.9%), conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs, 87.5%), or prednisone (46.9%). Importantly, medication use was not found to be associated with MCII in RA disease activity (Fig. 1), highlighting the urgent need for more reliable predictors of treatment response. The average interval between baseline and follow-up visits was 9.5 months. Disease activity varied across patients, with a mean CDAI of 16.3 at baseline and 13.6 at follow-up.
Figure 1 visually represents the medication usage among the 32 participants in the study. Statistical analysis using Fisher’s exact test revealed no significant correlation between the use of any specific medication and achieving MCII. This emphasizes the complexity of predicting clinical improvement in RA and the necessity to explore alternative biomarkers beyond medication regimens. bDMARDs stand for biologic disease-modifying anti-rheumatic drugs, csDMARDs for conventional synthetic disease-modifying anti-rheumatic drugs, and MCII for minimum clinically important improvement. MCII+ denotes patients who achieved MCII, while MCII− represents those who did not. N/A signifies data not available.
In this cohort, 37.5% of participants achieved MCII at the follow-up visit. The average CDAI change for these patients was -16.7 units, significantly different from the 5.7 unit average increase in CDAI for those who did not achieve MCII. Statistical tests (Fisher’s exact and Mann-Whitney U) were employed to identify factors associated with MCII. Baseline CDAI was significantly associated with the MCII patient group. At follow-up, factors significantly associated with MCII included CDAI, change in CDAI, pain (VAS), tender joint count (TJC), and patient and provider global evaluations of disease activity. Age was not significantly different between the MCII groups.
Gut Microbiome Composition Variance Explained by MCII Patient Group
To understand the role of the gut microbiome, PERMANOVA analysis was conducted to assess patient characteristics influencing gut microbial communities. The analysis considered MCII patient group, age, sex, smoking status, baseline CDAI, and medication use. It’s important to note that the variance explained by each factor in the adjusted model is assumed to be statistically independent.
The study revealed that the MCII patient group explained 3.8% of the variance in gut microbial communities (p=0.002, PERMANOVA; Table 2 and Fig. 2a), even after adjusting for other factors including age, CDAI, sex, smoking, and medication, as well as individual patient variations over time. Age, csDMARD use, sex, and smoking status also significantly contributed to the variance (7.7%, 3.1%, 2.9%, and 2.7% respectively), suggesting partial influence on microbiome profiles. However, CDAI, bDMARDs, and prednisone use were not significantly associated with gut microbiome composition. Subsequent analyses controlled for age, csDMARD use, sex, and smoking status to further investigate microbiome differences between MCII+ and MCII− groups.
Figure 2 showcases Principal Coordinates Analysis (PCoA) ordination plots of gut microbiome samples from RA patients (n=32). PERMANOVA analysis demonstrates that variance in gut microbial community composition is significantly explained by (a) MCII patient group, (b) age group, (c) csDMARDs use, (d) sex, and (e) smoking status, but not by (f) CDAI, (g) bDMARDs use, or (h) prednisone use. The analysis included 64 gut microbiome samples from 32 patients across both visits, accounting for intra-subject longitudinal variation by constraining permutations within visits. R² and P values are derived from adjusted PERMANOVA models. Circles and triangles represent baseline and follow-up samples, respectively, with lines connecting time points for the same patients. MCII, MCII+, and MCII− are defined as in Figure 1. Non-integer “n” values indicate instances where patient reports differed between baseline and follow-up.
Baseline Gut Microbiome Differences Between MCII Groups
Analysis of baseline gut microbiomes revealed Bacteroidetes and Firmicutes as the most prevalent phyla, Bacteroidales and Clostridiales as dominant orders, and Bacteroidaceae as the most abundant family. The study then investigated ecological diversity (alpha- and beta-diversity) and specific microbial taxa and metabolic pathways differing between MCII groups at baseline. This approach aimed to identify baseline microbiome features that could predict clinical improvement.
The MCII+ group exhibited higher species-level alpha-diversity (Fisher’s Index and richness) and beta-diversity compared to the MCII− group (Fig. 3). Specifically, six microbial taxa were more abundant in the MCII+ group: Negativicutes (class), Selenomonadales (order), Prevotellaceae (family), Coprococcus (genus), Bacteroides sp. 3_1_19 (species), and Bilophila sp. 4_1_30 (species). Conversely, Eubacterium sp. 3_1_31 (species) was more abundant in the MCII− group. Fifteen MetaCyc pathways also showed differential abundance. Pathways for tetrahydrofolate and L-methionine biosynthesis were enriched in the MCII+ group, while pathways for L-arginine, L-ornithine biosynthesis, and L-rhamnose degradation were more abundant in the MCII− group. These findings suggest that distinct gut microbiome profiles exist at baseline, even before clinical outcomes are evident.
Figure 3 illustrates the differences in baseline gut microbiome features between MCII+ and MCII− patient groups. (a) and (b) demonstrate significantly higher species-level alpha-diversity in the MCII+ group, measured by Fisher’s Index and richness respectively. (c) shows a higher distribution of Bray-Curtis distances in the MCII+ group, indicating greater beta-diversity. (d) highlights the seven microbial taxa with significantly different distributions, with six being more abundant in the MCII+ group. (e) identifies fifteen MetaCyc biochemical pathways differentially abundant between the groups. Statistical significance was tested using multiple linear regression models (MLRMs) for alpha-diversity and taxa/pathways, and Mann-Whitney U test for beta-diversity, controlling for age, sex, smoking, and csDMARD use. P values represent the regression model coefficient for the MCII patient group. *, 0.01 ≤ P < 0.05; **, 0.005 ≤ P < 0.01.
At follow-up, a significant difference in species-level Fisher’s Index remained between MCII groups. However, richness and beta-diversity did not show significant differences. Thirteen microbial clades differed significantly at follow-up, including Negativicutes, Bifidobacteriales, and Selenomonadales; Bifidobacteriaceae, Prevotellaceae, and Oscillospiraceae; Bifidobacterium and Veillonella; and Clostridium leptum and Roseburia inulinvorans. Only “Superpathway of Polyamine Biosynthesis II” was differentially abundant at follow-up pathway level.
Longitudinal Gut Microbiome Changes and Clinical Outcomes
The study further explored longitudinal changes in microbiome composition, examining fold-changes in microbial taxa and biochemical pathways from baseline to follow-up in relation to clinical improvement. This analysis aimed to identify how gut microbiome dynamics are associated with clinical outcomes.
Significant differences in fold-change were observed in eight microbial taxa between MCII groups (Fig. 4a). Gammaproteobacteria, Oscillibacter, Veillonella, and Bacteroides vulgatus showed higher fold-changes in the MCII+ group, suggesting increased abundance over time. Conversely, Coprococcus, Ruminococcus, Anaerotruncus colihominis, and Oscillibacter sp. KLE_1728 showed higher fold-changes in the MCII− group. Notably, changes in Gammaproteobacteria, Coprococcus, Oscillibacter, Ruminococcus, and Anaerotruncus colihominis often diverged in opposite directions between the two groups.
Figure 4 demonstrates significantly different fold-changes in microbial taxa and biochemical pathways between MCII+ and MCII− groups from baseline to follow-up. (a) highlights eight microbial taxa showing significant fold-change differences. For instance, Oscillibacter increased in relative abundance in the MCII+ group but decreased in the MCII− group, while Coprococcus, Ruminococcus, and Anaerotruncus colihominis showed opposite trends. (b) identifies seven MetaCyc biochemical pathways with significantly different fold-changes. P values, shown above box plots, were obtained using multiple linear regression models (MLRMs) to assess the association between MCII patient group and fold-change, controlling for age, sex, smoking, duration between visits, and csDMARD use. *, 0.01 ≤ P < 0.05.
Seven biochemical pathways also exhibited significant fold-change differences (Fig. 4b). Pathways related to sugar metabolism, such as rhamnose degradation and heptose derivative biosynthesis, had higher fold-changes in the MCII+ group. In contrast, pathways like “Superpathway of Aromatic Amino Acid Biosynthesis” and “Chorismate Biosynthesis from 3-dehydroquinate” had higher fold-changes in the MCII− group. Similar to taxa, fold-changes in pathways like ADP-L-glycero- and beta-D-manno-heptose Biosynthesis and Lipid IVA biosynthesis generally increased in MCII+ but decreased in MCII−, while others showed opposite trends. These diverging changes in specific microbiome features between groups may provide crucial insights into the relationship between gut microbiome dynamics and clinical improvement in RA.
Gut Microbiome as a Predictive Tool for Clinical Improvement and Disease Activity
Recognizing the challenge of predicting treatment response in RA, this study investigated the predictive capacity of the gut microbiome. Using baseline gut microbiome profiles, clinical, and demographic data, a neural network classification model was developed to predict MCII class (Fig. 5a).
The model achieved a high prediction accuracy in leave-one-out cross-validation, with a balanced accuracy of 90.0%. Notably, it correctly predicted MCII in all 12 patients who showed clinical improvement. The neural network outperformed other machine learning models, demonstrating its superior predictive utility.
Figure 5 evaluates the performance of neural network-based prediction models for MCII and CDAI. (a) illustrates the neural network model designed to classify patients into MCII groups using baseline data. (b) presents the confusion matrix from leave-one-out cross-validation, showing an overall classification accuracy of 87.5% and balanced accuracy of 90.0%. (c) ranks model input features by scaled importance, highlighting microbiome data as more influential than clinical/demographic information. (d) depicts a neural network model for predicting CDAI. (e) shows a scatter plot of observed vs. predicted CDAI scores, demonstrating a moderate correlation (Spearman’s ρ = 0.37, P = 0.003). The dashed violet line represents perfect prediction (y=x).
Feature importance analysis revealed that microbiome data, particularly taxonomic and functional components, were the most influential predictors of MCII (Fig. 5c). The top features included Sucrose Degradation III pathway, Parabacteroides sp. D25, Roseburia genus, Fatty Acid and beta-oxidation II pathway, and Biotin Biosynthesis I pathway. Clinical and demographic features ranked much lower, with csDMARD use being the highest ranked non-microbiome feature at 78th. This emphasizes the dominant role of the gut microbiome in predicting clinical improvement in this model.
Interestingly, the most important predictive features were not necessarily those showing significant differential abundance between MCII groups. This suggests that the neural network leverages complex, non-linear combinations of features, where even weakly associated features collectively contribute to strong predictive power.
Furthermore, a separate neural network model was developed to directly predict CDAI using the same input variables (Fig. 5d). This is a novel approach for chronic diseases. The model demonstrated a moderate but significant correlation between predicted and observed CDAI (Spearman’s ρ = 0.37, p=0.003; Fig. 5e). However, the model tended to under-predict higher CDAI values and over-predict lower values, potentially indicating a breakpoint in the relationship between predictors and CDAI around a score of 15.
In conclusion, this research, identified by 00957, strongly suggests that the gut microbiome holds significant promise as a non-invasive tool for predicting clinical improvement and potentially monitoring disease activity in Rheumatoid Arthritis. The study’s findings pave the way for future research to validate these predictive models and explore therapeutic interventions targeting the gut microbiome to improve RA patient outcomes.